Modelling gene expression profiles related to prostate tumor progression using binary states
Export citation
Abstract
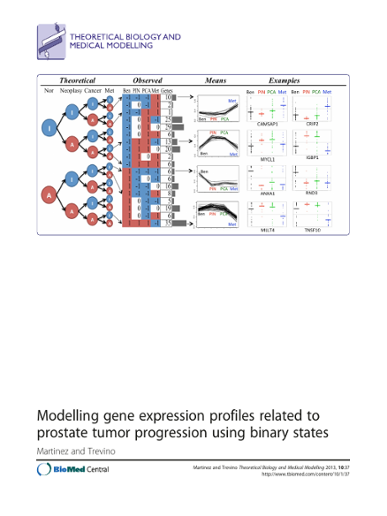
Abstract Background Cancer is a complex disease commonly characterized by the disrupted activity of several cancer-related genes such as oncogenes and tumor-suppressor genes. Previous studies suggest that the process of tumor progression to malignancy is dynamic and can be traced by changes in gene expression. Despite the enormous efforts made for differential expression detection and biomarker discovery, few methods have been designed to model the gene expression level to tumor stage during malignancy progression. Such models could help us understand the dynamics and simplify or reveal the complexity of tumor progression. Methods We have modeled an on-off state of gene activation per sample then per stage to select gene expression profiles associated to tumor progression. The selection is guided by statistical significance of profiles based on random permutated datasets. Results We show that our method identifies expected profiles corresponding to oncogenes and tumor suppressor genes in a prostate tumor progression dataset. Comparisons with other methods support our findings and indicate that a considerable proportion of significant profiles is not found by other statistical tests commonly used to detect differential expression between tumor stages nor found by other tailored methods. Ontology and pathway analysis concurred with these findings. Conclusions Results suggest that our methodology may be a valuable tool to study tumor malignancy progression, which might reveal novel cancer therapies.
Collections
- Artículo 1151



Except where otherwise noted, this item's license is described as http://creativecommons.org/licenses/by-nc-nd/4.0/